Your First Script¶
On HPC clusters, commands are rarely executed interactively due to shared resources and queueing policies. Instead, users prepare a batch script that describes the job requirements and execution steps. This script is submitted to the scheduler (Slurm), which runs it when the requested resources become available.
The most common way to submit a batch job to the scheduler is using the
sbatch command.
In this example, we submit a script called my_script.sh:
sbatch my_script.sh
SLURM reads the job requirements defined in the script, allocates suitable resources, and then launches the execution on the selected compute nodes.
Creating Batch Scripts¶
Job Builder
You can create your own batch script using our interactive Job Builder.
A batch script is typically organized into the following sections:
- Interpreter used to execute the script (e.g.
bash,python) - SLURM directives defining job parameters such as resources, run time, and partition
- Preparation steps such as loading modules, setting environment variables, or preparing input files
- Job execution where the application is launched
- Epilogue steps for post-processing, data movement, or cleanup
As an example, consider the following simple batch job script:
#!/bin/bash
#SBATCH -J "slurm test" # Job name
#SBATCH -N 1 # Request 1 node
#SBATCH --ntasks-per-node=16 # Run 16 tasks (processes) on that node
#SBATCH -o test.%J.out # File to write standard output (%J = job ID)
#SBATCH -e test.%J.err # File to write standard error
module load intel # Load required environment (compiler, MPI, etc.)
mpirun /bin/hostname # Run hostname command 16 times (once per task)
exit
This script defines only a minimal job requirements, such as number of nodes and tasks, standard output and error files and then runs
hostname command (which displays the server name) in parallel.
sbatch my_script.sh
Submitted batch job 38678
ls -ltr
total 8
-rw-rw-r-- 1 user user 198 Sep 21 14:08 my_script.sh
-rw-rw-r-- 1 user user 0 Sep 21 14:08 test.38678.err
-rw-rw-r-- 1 user user 80 Sep 21 14:08 test.38678.out
cat test.38678.out
n079
n079
n079
n079
n079
n079
n079
n079
n079
n079
n079
n079
n079
n079
As you can see, the job's ID was 38678 (displayed right after job submission). As expected, this number
was also used in STDERR and STDOUT file names. Since we requested 16 tasks on one node, the output file
contains 16 outputs of hostname command (and you can see the script was actually executed on n079).
Obviously, this is just a demonstration script and if you want to use it for a real HPC application, you have
to modify it accordingly. You can find more examples for specific applications in the
Devana Software or Devana Software
sections of userdocs portal.
Typical Batch Job Types¶
#!/bin/bash
#SBATCH -J serial_job # Job name
#SBATCH -N 1 # Use 1 node
#SBATCH -n 1 # Run 1 task (single-core)
#SBATCH -t 00:05:00 # Maximum runtime: 5 minutes
#SBATCH -o serial.out # Output file
#SBATCH -e serial.err # Error file
./my_serial_app
#!/bin/bash
#SBATCH -J openmp_job # Job name
#SBATCH -N 1 # 1 node
#SBATCH -n 1 # 1 task (OpenMP uses threads, not tasks)
#SBATCH -c 8 # Request 8 CPUs per task (i.e., 8 threads)
#SBATCH -t 00:10:00 # 10-minute wall time
#SBATCH -o openmp.out # Output file
#SBATCH -e openmp.err # Error file
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK}
./my_openmp_app
#!/bin/bash
#SBATCH -J mpi_job # Job name
#SBATCH -N 2 # Use 2 nodes
#SBATCH --ntasks-per-node=8 # 8 tasks (processes) per node (total: 16)
#SBATCH -t 00:15:00 # 15-minute time limit
#SBATCH -o mpi.out # Output file
#SBATCH -e mpi.err # Error file
module load openmpi # Load OpenMPI environment
mpirun ./my_mpi_app
#!/bin/bash
#SBATCH -J hybrid_job # Job name
#SBATCH -N 2 # 2 nodes
#SBATCH --ntasks-per-node=4 # 4 MPI tasks per node
#SBATCH -c 4 # 4 CPUs (threads) per task
#SBATCH -t 00:30:00 # 30-minute limit
#SBATCH -o hybrid.out # Output file
#SBATCH -e hybrid.err # Error file
export OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK} # OpenMP thread count
mpirun ./my_hybrid_app
#!/bin/bash
#SBATCH -J gpu_job # Job name
#SBATCH -N 1 # 1 node
#SBATCH --gres=gpu:1 # Request 1 GPU
#SBATCH -t 00:20:00 # 20-minute limit
#SBATCH -o gpu.out # Output file
#SBATCH -e gpu.err # Error file
module load cuda # Load CUDA environment
./my_gpu_program
Environment Setup¶
Many applications require a prepared software environment before they can run. This usually involves loading software modules, activating Python environments, or running programs inside containers.
Loading modules
module purge
module load gcc/12.1.0 openmpi/4.1.4
Using Conda
source activate myenv
Using containers
singularity exec my_container.sif ./my_program
Job Submission¶
At a minimum a job submission script must include number of nodes, time, type of partition and nodes (resource allocation constraint and features). If a script does not specify any of these options then a default may be applied.
Task distribution options
| Option | Description |
|---|---|
-a, --array=<index> |
Job array specification (sbatch only) |
--cpu-bind=<type> |
Bind tasks to specific CPUs (srun only) |
-c, --cpus-per-task=<count> |
Number of CPUs required per task |
--gpus-per-task=<list> |
Number of GPUs required per task |
--mem=<size>[units] |
Memory required per allocated node (e.g., 16GB) |
--mem-per-cpu=<size>[units] |
Memory required per allocated CPU (e.g., 2GB) |
--nodes |
Number of nodes to be allocated to the job |
--ntasks |
Set the maximum number of tasks (MPI ranks) |
-N, --nodes=<count> |
Number of nodes required for the job |
-n, --ntasks=<count> |
Number of tasks to be launched |
--ntasks-per-node=<count> |
Number of tasks to be launched per node |
Within a job, you aim at running a certain number of tasks, and Slurm allow for a fine-grain control of the resource allocation that must be satisfied for each task.
Beware of Slurm terminology in Multicore Architecture!
- Slurm Node = Physical node, specified with
-N <#nodes>- Always add explicit number of expected number of tasks per node using
--ntasks-per-node <n>. This way you control the node footprint of your job.
- Always add explicit number of expected number of tasks per node using
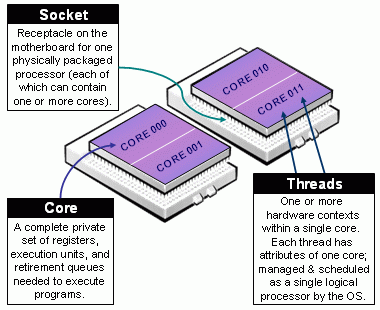
-
Slurm CPU = Physical CORE
- Always use
-c <threads>or--cpus-per-task <threads>to specify the number of cpus reserved per task. - Hyper-Threading (HT) Technology is disabled on Devana cluster.
- Always use
-
Assume cores = threads, thus when using
-c <threads>, you can safely setto automatically abstract from the job context.OMP_NUM_THREADS=${SLURM_CPUS_PER_TASK:-1}
The total number of tasks defined in a given job is stored in the $SLURM_NTASKS environment variable.
The --cpus-per-task option of srun in Slurm 23.11 and later
In the latest versions of Slurm srun inherits the --cpus-per-task value requested by salloc or sbatch by reading the value of SLURM_CPUS_PER_TASK, as for any other option. This behavior may differ from some older versions where special handling was required to propagate the --cpus-per-task option to srun.
In case you would like to launch multiple programs in a single allocation/batch script, divide the resources accordingly by requesting resources with srun when launching the process:
srun --cpus-per-task <some of the SLURM_CPUS_PER_TASK> --ntasks <some of the SLURM_NTASKS> [...] <program>
Basic accounting and scheduling options
| Option | Description |
|---|---|
-A, --account=<account> |
Charge resources used by this job to the specified user project. |
-e, --error=<filename> |
File in which to store job error messages (sbatch and srun only) |
--exclusive |
Reserve all CPUs and GPUs on allocated nodes |
-J, --job-name=<name> |
Job name |
--mail-user=<address> |
E-mail address |
-o, --output=<filename> |
File in which to store job output (sbatch and srun only) |
-p, --partition=<names> |
Partition in which to run the job |
-t, --time=<time> |
Limit for job run time |
A full list of variables that specify data handled by sbatch can be
found with the man sbatch command or by visiting the slurm documentation on
sbatch.
Common Tips
- Always request the minimum resources and time needed.
- Use
module purgeto prevent environment conflicts. - Use SLURM environment variables:
$SLURM_NTASKS,$SLURM_CPUS_PER_TASK - Define
OMP_NUM_THREADSproperly if using OpenMP. - Check
man sbatchor visit official Slurm documentation.
Debugging Failed Jobs¶
If a job fails, the following steps can help identify the problem.
- Check the
.errfile generated by the job for error messages. - Inspect job details using:
sacct -j <jobid>
scontrol show job <jobid>
- Configure email notifications to receive information about job completion or failure:
#SBATCH --mail-user=your@email.com
#SBATCH --mail-type=END,FAIL
- While debugging, submit short test jobs to reduce queue time:
#SBATCH -t 00:01:00
- For quick testing, run the application in an interactive allocation:
salloc -N 1 -n 4 --time=00:10:00
mpirun ./my_program
- If the job terminates with a non-zero exit code, increasing script verbosity may help identify the issue:
#!/bin/bash -x
Common Job Problems¶
The following table summarizes several common issues encountered when running jobs on the cluster and possible ways to resolve them.
| Problem | Possible Cause | Suggested Action |
|---|---|---|
Job stays in PD (Pending) state |
No resources available or project quota exhausted | Run squeue -j <jobid> to see the reason. Check project allocation using sprojects. |
| Job fails immediately | Error in the batch script or application command | Check the .err file generated by the job for error messages. |
| Job exceeds time limit | Runtime longer than the requested time | Increase the --time parameter in the batch script. |
| Job killed due to memory limit | Insufficient memory requested | Increase requested memory using --mem or --mem-per-cpu. |
| Job runs but produces no output | Incorrect input files or paths | Verify input file paths and working directory. |
| Application fails during execution | Environment not configured correctly | Ensure required modules or environments are loaded before running the application. |
For detailed inspection of a job, the following commands are often useful:
sacct -j <jobid>
scontrol show job <jobid>